What is sickle cell disease?
Sickle cell disease is the presence of haemoglobin S and any other abnormal haemoglobin. The haemoglobin is a protein contained in your red cells. It is responsible for transporting oxygen from tissues to the lungs and back (transporting and delivering oxygen around the body).
The haemoglobin is usually made of 2 major chains, 2 alpha chains and 2 beta chains. There are different types of haemoglobin variants with up to 1200 discovered so far, however, the normal haemoglobin is A. It is noteworthy too that not all of these haemoglobin types have clinical significance.
In newborn babies, there is usually haemoglobin F (also called fetal haemoglobin). However, as children grow into older infants, the fetal haemoglobin (haemoglobin F or HbF) production declines as the person begins to produce Haemoglobin A (or Hb A) which is the normal.
In sickle cell anaemia however, haemoglobin A is being replaced by production of haemoglobin S.
How and why abnormal haemoglobin is produced?
Our general body makes up, no matter how minute is decided by amino acid arrangement. Amino acids are the building blocks of proteins in our body. The amino acids in the DNA that determine how we appear or what we are made up of are very specific. So when the haemoglobin chains are being formed, the amino acid sequencing/arrangement on a particular chromosome is the determinant. What happens is that in valine replaces/takes the position of glutamic acid.
This singular mutation changes the type of haemoglobin the body produces from A to S. There are other types of haemoglobin too depending on what displacement occurs. A second example is when lysine, another amino acid replaces glutamic acid on that sequence, it leads to the production of yet another haemoglobin, which is haemoglobin C. Other forms of sickle cell disease (other sickle cell syndromes) include:
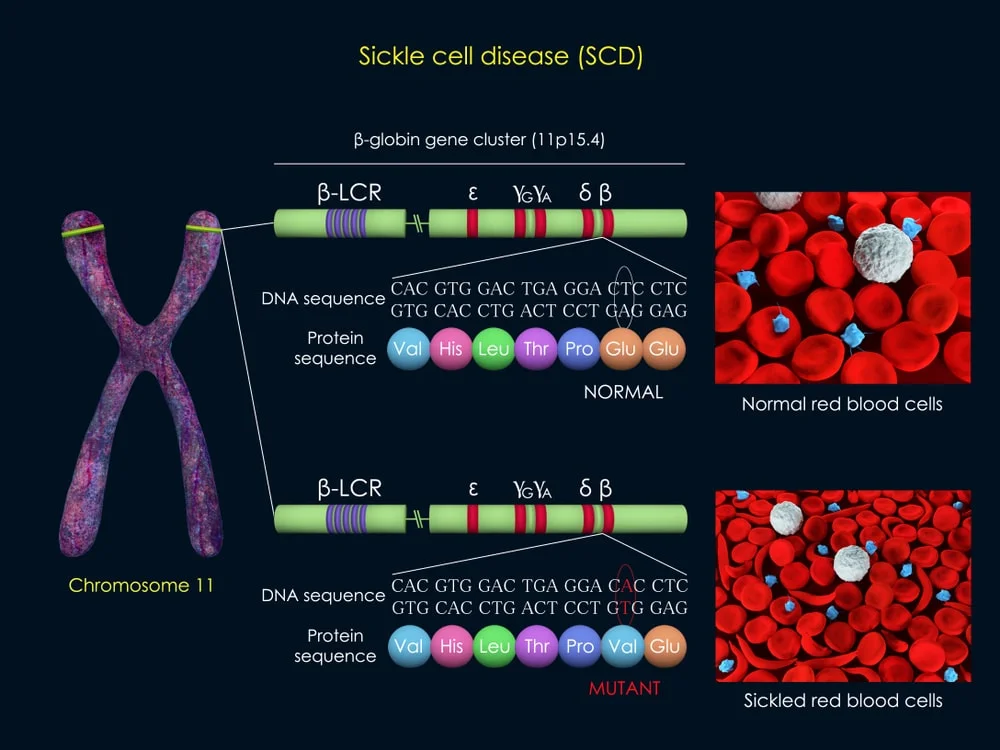
Hb S-beta thalassemia, HbSC, HbS-O Arab, sickle cell with alpha thalassemia, sickle cell anaemia with high persistence of fetal haemoglobin (HbSS with HPFH).
. Those with Hb- SS however suffer the severest form of the disease.
Whenever a person produces haemoglobin S and another abnormal haemoglobin, we say they have sickle cell disease (whether it is SS, SC, SO-Arab, S+beta –thalassaemia). Whenever they have homozygous disease (both S as in HBSS or SS) we say it is sickle cell anaemia.
Mode of inheritance
Sickle cell disease is an inherited genetic disorder, inherited in an autosomal recessive pattern meaning that sufferers can only inherit it from two parents who have traits of the disease. Those who have traits are those who have haemoglobin AS. They do not show any clinical symptoms and live normal lives. However, if 2 people who have the traits have biological children together, they have a 25% chance per pregnancy of producing a child with sickle cell disorder.
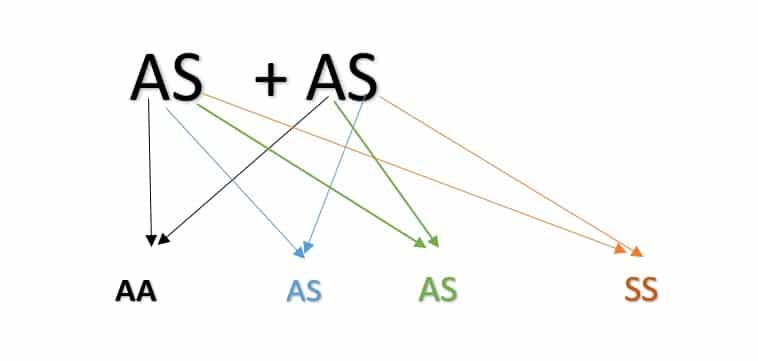
The above picture shows possible outcome of genotypes per pregnancy. There is no method to currently predict how the body decides which haemoglobin the baby will be born with. For the above combination, AA- 25% per pregnancy, AS- 50%, SS-25%. In no particular order. It means that couples who are both carriers can have all AA or all AS or all SS or any combination.
The sickle cell haplotypes
Sickle cell anaemia can have varying haplotypes meaning that there could be DNA variations in the inheritance pattern which may affect the clinical outcome. In other words, some haplotypes have milder clinical manifestations than others. Haplotypes are named after areas they are said to have originated from. Haplotypes known include:
- The Benin haplotype
- The Senegal haplotype
- The Bantu haplotype
- The Arab-Indian/Saudi Arabia-Indian haplotype
- The Cameroon haplotype
Why is abnormal haemoglobin a problem?
The haemoglobin is found in the red cell. Its main function is to carry oxygen. When the normal haemoglobin (haemoglobin A) is produced, transportation of oxygen proceeds smoothly. The red cell is also able to maintain its normal shape despite stressful conditions such as deoxygenation, dehydration. The red cells are also able to pass through microvasculature (small vessels) and maintain their biconcave shape.
The shape of the normal red cell is biconcave, (like a doughnut not with a completely hollow middle but with a thin depressed middle). This shape ideally should be maintained whether the red cell is carrying oxygen at that moment or not.
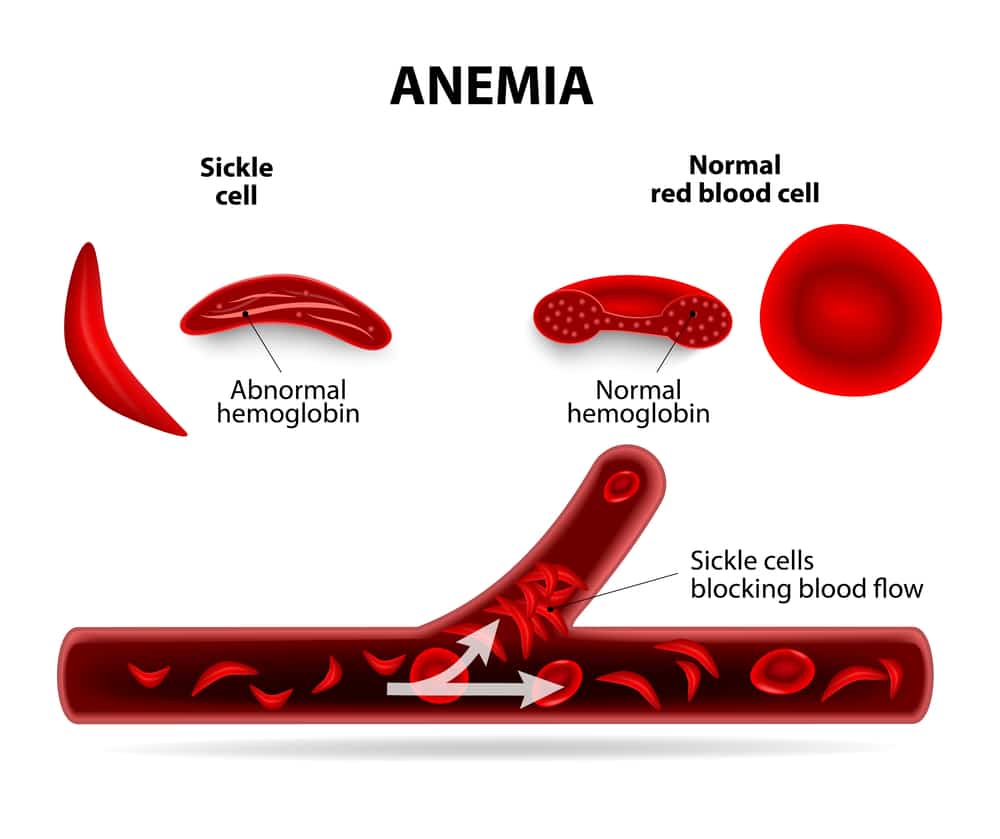
However, when abnormal haemoglobin is produced, the red blood cell is not able to maintain its shape after a little stress already mentioned. It gets easily denatured and becomes sickle-shaped hence the name ‘sickle cell’. This shaped does not allow the red cell function fully in carrying oxygen around the body. It also shortens the life span of the red cells reducing it from 120 days to 10-30 days. The sickle-shaped cells are easily destroyed because there is a system in our body led by the spleen designed to destroy dysfunctional blood cells.
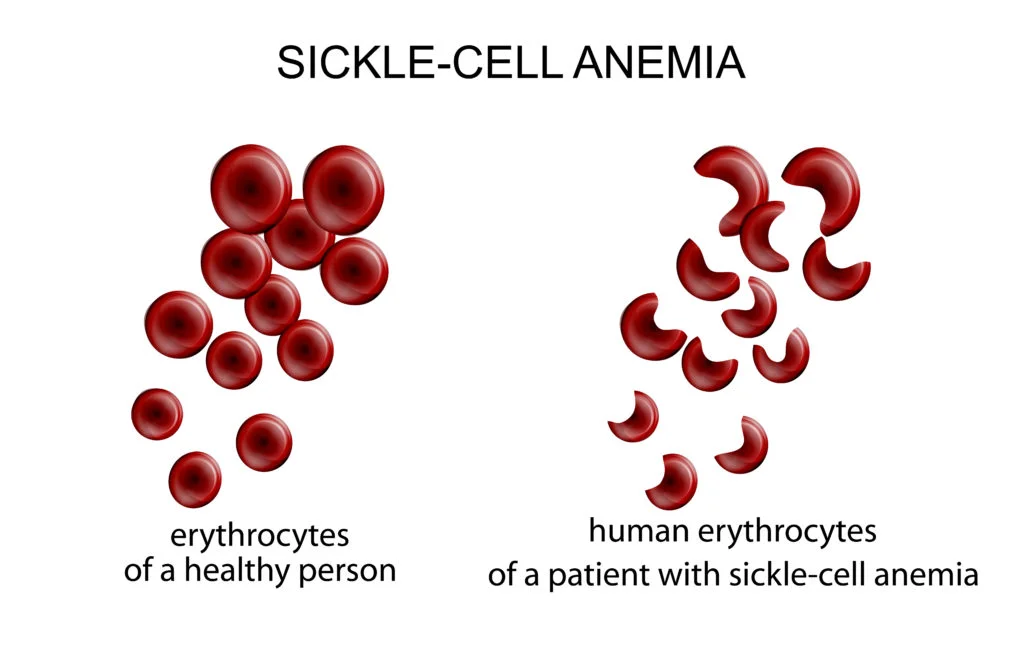
The abnormally shaped red cells also begin to form stacks and obstruct the microvasculature (the small tube-like vessels that through which blood flows example arterioles and venules). This obstruction can be imagined/envisioned, like accumulating debris in a pipe. This retards blood flow and subsequently leads to the hallmark of acute sickle cell presentation known as the vaso-occlusive crisis. Vaso occlusion means blocking of the vessels.
So basically, this genetic aberration leads to abnormal red cell shape, reduced oxygen-carrying capacity short lifespan due to destruction, acute/emergent presentations and later, chronic complications.
Haemoglobin F (HbF /fetal haemoglobin) is the haemoglobin type babies are born with, however, even though it is not haemoglobin A, it gives protection to infants because of its high affinity to oxygen, meaning it binds to oxygen more readily which is advantageous. However, as babies grow older, they begin to lose haemoglobin F which is then replaced by other haemoglobin types examples A, S. When babies are born, fetal haemoglobin (HbF) can get as high as 85% and can fall as low as less than 1% at the child’s first birthday.
Normal value expected in adults ranges between 0.8-2 %. Some people who live with sickle cell anaemia may have high levels of HbF which gives them a better clinical outcome. Some have their HbF levels as high as 30% (normal level is 0.8-2% in adults) and usually referred to as HbF.
Epidemiology of sickle cell disease
Sickle cell disorder belongs to a group of genetic disorders of haemoglobin commonly called haemoglobinopathies. Sickle cell disorder is common among Africans, African-Americans, people from the middle east and Indian subcontinent, Caribbean.
The sickle cell gene is said to be in West Africa as a protection against falciparum malaria, however, people with sickle cell disorder began to emerge as carriers began to reproduce.
Signs and symptoms of sickle cell disease
Due to the presence of haemoglobin F, infants are usually protected from sickling until much later. However, some may begin to show symptoms as early as 6 months of age. Sickle cell disorder is characterized by the presence of acute events popularly known as crises and chronic complications. The bedrock of these events are due to the vaso-occlusion which comprises of painful episodes (obstruction of blood vessels) and haemolysis (breakdown of red cells).
Clinical features (Signs and symptoms)
- Sickle cell habitus
- Acute events and crisis
- Chronic complications
Sickle cell habitus
Habitus is the general outlook of a person especially their physical appearance. Many who live with sickle cell disorder have a general outlook we refer to as the sickle cell habitus. These features result from activities that certain organs have to undergo because of the disease.
- Bossing (protuberance) of the skull and maxilla with protruding teeth.
- Pallor- the eyes may be pale
- Short stature
- Jaundice- yellowness of the eyes.
- Long upper and lower limbs which may not be proportionate with the rest of the body.
- Shortened fingers or limbs depending on complications already suffered
- There may be protuberant abdomen
Acute sickle cell events and crises
Triggers of crisis
Usually, crises in sickle cell disease are usually triggered by a number of factors
- Dehydration
- Infection
- Stress
- Physical activity
Vaso-occlusive crisis
It is the most common presentation experienced in this disorder and the commonest emergency. The sickle-shaped red cells obstruct the small vessels and so blood supply does not get to organs. This is followed by pains with varying severity and can happen on any part of the body especially bones and joints. The recurrent vaso-occlusion continue to decrease blood supply to organs.
Those who have the sickle cell trait (AS) live normal lives and do not show any symptoms ordinarily. However, they may experience some symptoms in certain extreme conditions of dehydration.
Hyperhaemolytic crisis
Sickle cell anaemia is already a chronic haemolytic state (there is constant breakdown of red blood). However, sufferers can present in the emergency with symptoms such as passage of very dark urine, yellowness of the eyes following certain triggers like infections. These features stated suggest an active increased rate in the destruction of red blood.
Sequestration crisis
It is a fatal emergency involving the sudden trapping of blood. It usually occurs in the spleen and sometimes in the liver. It is commoner in children. There is a sudden step wise drop in the packed cell volume or haemoglobin level (blood level) and persons become pale. If blood is being sequestered by the spleen, there is usually a palpable enlargement of the spleen with left upper quadrant pain (left upper part of the abdomen just below the ribs) and if sequestration occurs in the liver, there is enlargement of the liver with right upper quadrant pain (right upper part of the abdomen just below the ribs).
Aplastic crisis
This crisis involves sudden suppression in bone marrow function. The bone marrow is where your blood cells are originally produced. With this crisis, there is a general reduction in the production of blood cells generally. This crisis is caused by parvovirus B19 infection.
Other acute events
Hand-foot syndrome (dactylitis)
It is said to be one of the earliest features seen in children with sickle cell disorder. There is swelling of the hands and feet, very painful. It is common between the ages of 6 months to 3 years. Bone pain usually due to infarction to the bone marrow. As people grow older, crises involve more bones including the spine.
Acute chest syndrome
Is an emergency and very fatal. It is said to be currently the most fatal acute complication of sickle cell disease. Those with this syndrome will present to the hospital with fever, cough, difficulty in breathing, chest pain. It is usually preceded by a reduction in blood supply to the ribs following vaso-occlusion and/or chest infections, especially in children.
Acute abdominal crisis
When blood supplying the gut is trapped in sickle cell disease, the abdomen can become very painful, swollen, tense with a reduction in function. Usually, bloating, constipation (reduction in the usual passage of stools) will be experienced. This also involves a reduction in blood supply to intraabdominal organs like the liver, spleen, intestines. Continuous infarction can lead to the formation of abscesses, the formation of stones in the gall bladder and death of these organs.
Recurrent vaso-occlussion to the spleen leads to infarction (obstruction of blood supply leading to death of a tissue) and a process called auto splenectomy (self-removal of spleen).
Priapism
In males, there can be a persistent, painful, spontaneous penile erection without sexual desire and could lead to erectile dysfunction if not managed properly. Usually, we say it is priapism if there is unprovoked erection has exceeded 3-4hours.
Acute cerebral infarction/stroke
Stroke is a common emergency in sickle cell anaemia especially in the young and has a tendency to reoccur. This is the reason children with sickle cell disease between 2-16 years are placed on routine transcranial doppler scans. These scans are able to assess who is at higher risk and therefore guides the clinical decision of managing a team in preventing stroke.
Cholelithiasis (gallstone formation)
Normally, our body breaks down red blood cells after about 120 days and produces a by-product called bilirubin. However, in sickle cell disease, because of the constant breakdown, excess of this product is formed, it then accumulates leading to the formation of stones in the gallbladder. Usually, these stones block pathways in the gallbladder called ducts.
Renal papillary necrosis
Low oxygen levels and the formation of sickle cells can affect kidney function. So they may present with the passage of blood in urine because the sickled cells obstruct blood flow leading to the red blood finding its way out of the blood vessels into the path where urine is collected and coming out as blood in the urine or even cause the death of parts of the urine collecting system. This complication is commonest in HbSC.
Septic arthritis- those with sickle cell disorder are prone to infections including infections of joints by certain bacteria.
Chronic complications of sickle cell disease
The following are the long term effects of sickle cell disease on various organs.
The eyes
Retinopathy- this results from recurrent obstruction of blood vessels supplying the retina, leading to poor vision
The skin and skeletal system
Shortening of fingers can occur especially for those who recurrently who suffered hand-foot syndrome in childhood
Growth- children with sickle cell disorder may experience slowing of growth beginning after infancy. They may also experience delay in sexual maturation including commencement of menstruation in female adolescents.
Chronic leg ulcers- these are common complications where they have a long standing open injury on the skin (especially the legs) and very difficult but possible to treat.
Avascular necrosis of the head of humerus and femur- the humerus and femur are the long bones of arm and thigh. Due to recurrent obstruction of blood supply, the high end of these long bones known as the head can die from poor blood supply and often needs surgical intervention.
Osteomyelitis- recurrent obstruction of blood supply to bones leads to poor blood supply and spread of infections on the bone which may involve certain layers of the bone, bone marrow and soft tissue. Any bone can be affected.
The abdomen
Sickle cell hepatopathy- sickle cell predisposes the liver to many injuries resulting from occlusion of its blood supply, sequestration (trapping of blood) and formation of stones in the gall bladder and inflammation leading to reduce the function of the liver/liver disease.
Autosplenectomy- recurrent obstruction of blood supply to the spleen causes death and self-removal of the spleen
Sickle cell Nephropathy- this is a chronic condition resulting from recurrent obstruction of blood flow and decreased oxygen to the kidneys.
Sickle cell disease in pregnancy- usually in healthy individuals, there is a fall in haemoglobin levels and so pregnant women with sickle cell disease will experience reduction in haemoglobin level and they may experience more of the painful crisis after 28 weeks of pregnancy.
Cardiovascular system
Cardiopulmonary complications
The recurrent fall in red blood cells puts more work on the heart in a bid to meet up with body needs, the constant breakdown of red cells and its effect on blood vessels, obstruction of blood vessels by blood clots, reduction of oxygen supply all affect the heart and surrounding large vessels. There is a decrease in the function of the heart muscles, an increase in pressure of pulmonary vessels.
Determinants of clinical manifestations
Why do some people with sickle cell disease have a more severe disease than others?
- HbSS and HbS + beta thalassaemia cause more severe disease
- Haplotype- Some haplotypes usually more associated with severe clinical outcomes compared to other haplotypes. The Senegal haplotype is said to be associated with the mildest disease due to the presence of higher HbF.
- Presence of HbF- because of affinity for oxygen, there is a milder clinical disease with an increased level of HbF (fetal haemoglobin).
- Nutrition and socio-economic status are important factors in determining clinical outcome.
Diagnosis of sickle cell disease
Any of these tests will be performed to determine one’s genotype for the first time. However, methods that reveal the level of fetal haemoglobin may be used subsequently by the clinician before the commencement of certain medications. A full blood count will also be performed routinely.
- A complete blood count with peripheral blood film and red cell indices inclusive gives us a picture of the state of blood cells in circulation. For the two most common forms of sickle cell disease in West Africa, HbSS or HbSC, their appearance on blood films are different.
- Sickling test- used in diagnosing sickle cell disorder though not currently used by many modern centres.
- Haemoglobin electrophoresis- this helps identify common haemoglobin variants based on charges.
- High performance (pressure) liquid chromatography (HPLC)- shows us different haemoglobin variant as well as quantify them including fetal haemoglobin. It is a more modern method than haemoglobin electrophoresis and quantifies haemoglobin.
- Isoelectric focusing
- DNA Molecular testing can detect the mutation responsible for the disease. It is expensive and currently the most accurate.
It is important to note that with advancements in testing, we are able to pick more of the sickle cell syndromes, thalassaemias and foetal haemoglobin. Overtime time, tests have evolved to quantification/quantitation of haemoglobin. We have more centres now using HPLC as their basic screening because it quantifies haemoglobin unlike the first 3 tests stated above.
Treatment of sickle cell disease
- First aid
Before getting to the hospital, during a painful crisis, patients should be kept warm especially as extreme cold can trigger crisis, there should be adequate rehydration, warm towels can be placed on painful sites
- Acute events and crisis
They are usually managed as emergencies. Common treatment in the emergency room will include rehydration, analgesia and treatment of underlying cause for vaso-occlusive crisis. There may also be need for oxygen, blood transfusion, exchange blood transfusion, urgent CT scan depending on crisis, urological procedures etc. Interventions will be carried out according to diagnosis. It is important that those with crisis go to the emergency room on time as some emergencies have dire or irreversible consequences.
Multidisciplinary care
Sickle cell disease affects every system and so many sufferers will need to visit different specialities at different times depending on their clinical outcome.
Many complications will require multidisciplinary care such as haematologists being the primary physicians, the obstetricians, gastrointestinal surgeons, orthopaedic surgeons, clinical psychologists, neurologists, ophthalmologists, plastic surgeons, nephrologists, urologists, cardiologists, nephrologists.
General care and prevention of sickle cell disease
Those who live with sickle cell disorder must:
- Attend routine clinics and keep appointments with their doctors
- Undergo routine investigations. Most common is the full blood count and other investigations for liver function, renal function as the clinician decides.
- Ensure immunization according to local protocols for immunization
- Take their prescribed routine medications daily as this helps their general wellbeing by controlling anaemia, preventing malaria, help fight infections.
- Wear protective clothing, use insecticide treated nets especially for malaria endemic regions as malaria is a common trigger for crisis.
- Be protected from extremes of weather as this can trigger crisis.
- Ensure adequate water intake and balanced, healthy diet. Children with sickle cell disorder will need to be fed more number of times than children without the disorder.
Medication and sickle cell disorder
Ideally, routine medications are given to prevent malaria, help control anaemia etc. However, those who live with the disorder must stick to prescribed medications as certain analgesia can become addictive and detrimental. There are also current medications given to increase fetal haemoglobin levels, however, clinicians prescribe according clinical outcome including frequency of one’s vaso-occlusive crisis and chronic complications.
Cure of sickle cell disorder
Stem cell transplantation
This is a cure for sickle cell disease where stem cells are taken from matching AA donors and transplanted into those with sickle cell disease. Haemopoietic stem cells are the original cells from which blood cells originate. They are found in the circulation, the bone marrow or the umbilical cord.
In stem cell transplant, certain drugs are used to wipe-off the bone marrow of the recipient (the receiver) and the stem cells of a donor (giver) with AA genotype is transfused into the recipient. Donors can be siblings who must be genetically matched with recipient. Stem cell transplant so far has been more successful in children as the incidence of graft-versus-host-disease (an adverse reaction when the donor’s immune cells attacks the recipient’s tissues) is less. This reaction is more likely with increasing ages of both the donor and recipient.
Other complications could be infections, severe bleeding, failure of transplant, malignancies, autoimmune disease, cataracts.
New circulating blood cells may begin to emerge1-3 weeks after the procedure which is a sign of successful engraftment, recipients suffer severe immunodeficiency in the first 12-52 weeks following the procedure and their bone marrows reserve may be impaired for up to 24 months. The recipient’s blood group also changes to become that of the donor. Recipients will require post-transplant care and follow up.
Stem cell transplant can also be used to cure other diseases like leukemias, multiple myelomas.
Gene therapy
This is still being studied and it involves insertion of a gene to correct the mutation.
World sickle cell day and sickle cell as a global emergency
In December 2008, the United Nations general assembly recognized sickle cell as a public health problem and named June 19th world sickle cell day
Myths and common question about sickle cell diseases
Some general misconceptions/questions about sickle cell disease;
- That they are cured automatically after 21 years of age. This is not true.
- That if 2 carriers bear biological children together, they will automatically have 1 AA, 2 carriers and only the 4th will have sickle cell disease, that is not true. The truth is that there is a 25% chance per pregnancy of having a child with sickle cell disorder.
- That they never exceed 40 years of age. That is not true as we have them living into 6th, 7th decades of life.
- Do we give iron supplements or not? We do not routinely give iron medications to those who live with sickle cell disorder because of the risk of iron overload. This is due to the fact that the constant breakdown of red blood, as well as recurrent transfusion, may lead to iron overload. However, for those who are pregnant, their obstetricians may decide to give iron after proper clinical assessment. Iron overload is dangerous and can damage certain organs such as liver, adrenal glands, thyroid etc.
- Is there a way to find out the genotype of an unborn child? Yes, before 13 weeks of pregnancy, a procedure called chorionic villous sampling can be carried out to find out.
- Why do haematologists refer to haemoglobin SS as haemoglobin S and haemoglobin AA as haemoglobin A? this is because of the appearance on a popular test call haemoglobin electrophoresis. The results appear as bands so S is just one band, A is just another band. They do not appear as 2As or 2Ss.
Have a comment or want to learn more about sickle cell disease, join the sickle cell discussion.